
Junctional Epidermolysis Bullosa (JEB) is a rare and often severe form of EB caused by mutations in genes that affect the anchoring of the outer layer of skin (epidermis) to the underlying layer (dermis).
Blistering in JEB occurs within the lamina lucida, a portion of the basement membrane zone. Symptoms are typically present at birth and can range from mild to life-threatening. The severity varies considerably across the two major subtypes, intermediate and severe.
While some individuals with JEB may experience skin fragility and blistering mainly in response to trauma, others may have extensive wounds, mucosal involvement, and systemic complications that can significantly impact overall health and, in severe cases, may shorten life expectancy. Navigate to a specific section by clicking the links below:
"Brooklyn was born missing some skin on the bottom of her right foot and some nails, while the ones she had appeared somewhat discolored and differently shaped. She underwent genetic testing, and a week or two later, we found out that she has JEB with an LAMA3 gene mutation (two variants)."
Morgan, Mom to Brooklyn, Living with Junctional EB
Signs and Symptoms
Junctional Epidermolysis Bullosa (JEB) is characterized by recurring blistering of the skin, which can occur even with minimal friction or trauma. The severity of symptoms varies widely among subtypes, ranging from moderate skin fragility to more severe forms involving serious internal complications. Hallmark signs and symptoms of JEB include:
- Skin Fragility and Widespread Blistering: Blisters may form spontaneously or from minimal contact, particularly in areas exposed to pressure or friction.
- Chronic Wounds and Delayed Healing: Open wounds can be slow to heal and prone to infection, significantly affecting quality of life.
- Mucosal Involvement: Blistering may affect mucous membranes inside the mouth, nose, throat, eyes, and sometimes the gastrointestinal or genitourinary tract. Eye involvement can lead to corneal damage, scarring, and vision issues.
- Nail and Dental Abnormalities: Nails may be absent, thickened, or abnormally shaped. Enamel defects may increase the risk of dental issues.
- Hair Loss (Alopecia): Scalp blistering and chronic injury may cause scarring and permanent hair loss in some cases.
- Feeding and Nutritional Challenges: Oral and esophageal blistering may make eating painful, difficult, or sometimes impossible, requiring specialized diets or feeding tubes.
- Growth Delay and Failure to Thrive: Poor weight gain and slowed growth are common due to chronic wounds, nutritional difficulties, and increased energy demands.
- Breathing or Voice Changes: In some subtypes, involvement of the airway can cause respiratory issues; or voice box (larynx) can cause a hoarse cry or respiratory issues in infancy.
- Life-Threatening Complications: In the most severe form of JEB, extensive internal and external blistering can lead to life-threatening infections and complications early in life.
Subtypes of Junctional EB
Junctional EB includes two main subtypes, with several less common subtypes, categorized by the specific genetic mutations involved and the clinical features observed. These include:
JEB Intermediate
(Previously known as non-Herlitz or JEB generalized intermediate)
JEB Intermediate is characterized by blistering from birth and may include early-onset internal complications. Both JEB Intermediate and JEB Severe result from the same underlying genetic mutation, which disrupts the production of anchoring proteins that bind the skin layers together. The term “Intermediate” refers to the partial expression of these proteins— which allows for some production of these proteins allows resulting in a less severe involvement compared to JEB Severe. There is a wide spectrum of severity within the JEB Intermediate sub-type itself. While some experience an improvement with age, significant skin fragility, chronic wounds, and potential internal involvement may be present, requiring lifelong medical management.
Specific Features
Recurring blistering that often results from friction or minor trauma. Blisters tend to rupture leaving erosions, which can become extensive. Areas of ulcerated skin may be present at birth, most commonly on the lower limbs or dorsa of the feet and ankles. Blisters and ulcers may heal with atrophic scarring and variable hypo- or hyperpigmentation. Blistering may improve with age.
Blisters can appear in mucosal surfaces such as the mouth and nose.
May experience blisters in the eyes or on the conjunctiva, leading to irritation, dryness, scarring and potential visual impairments.
Scalp blistering may cause patchy or permanent hair loss starting in childhood.
Enamel defects that lead to a higher risk of dental problems.
Thickened, absent, or misshapen nails.
Most commonly presents in older individuals with urethral stricture disease.
Development of aggressive cutaneous squamous cell carcinoma (SCC) can occur in adulthood.
JEB Severe
(Previously known as JEB Herlitz or JEB generalized severe)
JEB Severe is characterized by widespread blistering, accompanied by profound internal complications. Both JEB Severe and JEB Intermediate share the same underlying genetic mutation, which disrupts the production of vital proteins. In JEB Severe, there is little to no functional production of these proteins, leading to life-threatening complications. Infants with JEB Severe face a significantly shortened life expectancy, with survival often not extending beyond the first two years of life.
Specific Features
Blisters may or may not be present at birth. In the early weeks of life, blisters may be few and tend to occur on the buttocks, elbows, and around the nails. By a few weeks to months of age, wounds may become chronic with friable granulation tissue, commonly affecting the face, ears, and distal digits. Persistent large gluteal wounds are common. Blisters also often rupture, leaving erosions that can become extensive.
Areas of ulcerated skin may be present from birth, and are most commonly found on the lower limbs, feet, and ankles. Blisters and ulcers can heal with atrophic scarring, leading to hypo- or hyperpigmentation.
Ocular involvement with corneal blistering and erosions. Pannus formation, scarring and symblepharon may follow episodes of blistering.
Blistering, erosions, granulation tissue, and scarring commonly affect the laryngeal and upper airway mucosa. This can cause hoarseness, stridor, and difficulty breathing.
Progressive airway narrowing may lead to life-threatening obstruction, sometimes requiring interventions like tracheostomy. Recurrent infections, aspiration, and respiratory failure are also common due to compromised mucosal integrity.
Infants usually develop profound failure to thrive despite adequate nutritional intake. Potential life-threatening internal issues, including respiratory complications, and organ involvement, often lead to sepsis or other life-threatening infections.
Other Junctional EB Variants
In addition to Junctional Intermediate and Junctional Severe, there are several less common and distinct subtypes that present with unique challenges.
Noted Clinical Symptoms
- Full thickness skin loss over extensive areas of the head, trunk and limbs at birth.
- Subsequent severe skin fragility.
- Skin loss can cause deformity of structures such as the ears and nose. Severe phenotypes can present with rudimentary ears.
- Nail dystrophy and loss.
- Pyloric atresia is usually evident within the first days-week of life.
- May have atresia at other gastrointestinal sites e.g. duodenal or anal.
- Usually lethal within the first few weeks of life despite surgical correction of pyloric atresia.
- Milder, non-lethal forms have less severe skin and nail involvement but with frequent genitourinary tract involvement.
Genetics
- Biallelic loss-of-function, splice site or missense pathogenic variants in ITGA6 result in a severe and rapidly lethal phenotype.
- Biallelic loss-of-function or splice site pathogenic variants in ITGB4 that result in loss of 4 integrin result in a severe and rapidly lethal phenotype.
- Biallelic loss-of-function, missense, splice site or in-frame deletion mutations in ITGB4 which result in reduced 4 integrin expression result in a milder form of JEB with pyloric atresia.
- Biallelic mutations in ITGB4 may result in JEB without pyloric atresia.
Noted Clinical Symptoms
- Limited cutaneous fragility and blistering, often only acral.
- Variable nail dystrophy and mucosal involvement.
- Variable dental enamel defects.
- Normal hair.
Genetics
- Autosomal recessive inheritance.
- Homozygous or compound heterozygous pathogenic variants in COL17A1, LAMA3, LAMB3, LAMC2, ITGB4, ITGA3.
Noted Clinical Symptoms
- Onset of blistering from birth in flexural sites.
- Atrophic scarring.
- Dental enamel abnormalities.
- Variable nail loss.
Genetics
- Pathogenic variants associated with residual expression of laminin 332.77
Noted Clinical Symptoms
- Onset of skin fragility in childhood often starting acrally.
- Progressive fragility with age.
- Healing with skin atrophy and loss of dermatoglyphs.
- Scarring leading to flexion contractures of the fingers and reduction of mouth opening may occur with age.
- Variable dental enamel and nail involvement.
Genetics
- Autosomal recessive inheritance.
- Biallelic pathogenic variants in COL17A1, specifically the missense variant c.3908G>A, p.R1303Q, cause this phenotype.
Syn. Shabbir syndrome.
Noted Clinical Symptoms
- Onset of skin fragility from birth with blistered areas leaving erosions and granulation tissue (much more than JEB severe).
- Predilection for the face and neck.
- Nail dystrophy and loss with granulation tissue of the nail beds.
- Conjunctival and eyelid granulation tissue leading to symblepharon, scarring and impaired vision.
- Laryngeal granulation tissue leading to respiratory obstruction which can be lethal.
- Profound Anaemia is a major feature due to bleeding from over granulating wounds.
Genetics
- Autosomal recessive inheritance.
- Most cases in Punjabi Muslim individuals with a homozygous founder single nucleotide insertion mutation in exon 39 of LAMA3 which is specific to the LAMA3A isoform.
- Compound heterozygosity for mutations in LAMA3A and LAMA3 result in a similar JEB-LOC phenotype.
syn. ILNEB, interstitial lung disease, nephrotic syndrome and epidermolysis bullosa
Noted Clinical Symptoms
- Variable cutaneous features with absence or presence of skin fragility from infancy.
- Nails may be dystrophic and hair may be sparse.
- Interstitial lung disease and nephrotic syndrome predominate the phenotype, and can be diagnosed soon after birth.
- Death in infancy or early childhood is the norm.
Genetics
- Most reported cases result from homozygous loss-of-function pathogenic variants in ITGA3.
- Missense ITGA3 mutations reported in milder cases surviving to later childhood.
Causes & Inheritance
Junctional EB is a genetic condition, caused by mutations in genes responsible for producing proteins that anchor the layers of skin together—most commonly LAMA3, LAMB3, LAMC2, and COL17A1. These proteins help form the structures that keep the epidermis attached to the dermis. When these proteins are missing or dysfunctional, the skin becomes fragile and prone to separation and blistering.
Most forms of JEB are inherited in an autosomal recessive pattern. This means a child must inherit two mutated copies of the gene (one from each parent) to be affected – each parent being a carrier of the recessive gene. Parents who are carriers thereby have a 25% chance with each pregnancy of having a child with JEB.
JEB is not contagious and cannot be caused or prevented through behavior or environmental changes—it results from genetic mutations occur at conception.
Impact on Daily Life
Junctional EB can greatly impact daily living. For some, this may mean ongoing wound care and managing skin fragility. For others—especially those with severe subtypes—chronic pain, nutritional support, frequent infections, and hospitalizations may be part of daily life. Families often manage complex care routines, including bandaging, specialized feeding, and respiratory support.
Despite these challenges, many individuals with JEB and their caregivers build strong support systems and find ways to adapt to their daily routines. Advances in care, early interventions, and improved understanding of JEB have led to a better quality of life.
For families impacted by the prognosis of JEB Severe, the journey may involve not only managing medical complexities but also coping with the emotional toll of a shortened life expectancy. Support networks, palliative care, and counseling can be crucial to navigating these challenges, offering both physical and emotional comfort during an incredibly difficult time.
The impact of JEB can vary widely depending on severity and presentation, with each person’s experience with EB being unique. Continue to explore our website for more detailed information on care and resources, and visit EBConnect.org, our online community platform, for valuable guidance and peer support.
Treatment and Care
Medical management of Junctional EB is largely supportive, focusing on wound care, pain management, and treating secondary complications. Learn more about EB care strategies and practical information on our How To Care section.
Healthcare professionals can also refer to our Emergency Management page for clinical guidance in urgent situations.
Emerging Treatments and Clinical Trials
In an incredible achievement, the U.S. FDA approved the first treatment for Junctional EB in 2023.
FILSUVEZ®, developed by Chiesi Global Rare Diseases, is a prescription topical gel that supports wound healing in adults and children 6 months of age and older. Learn more
Ongoing research is actively exploring promising therapies, and clinical trials play a critical role in advancing the understanding of Junctional EB and developing new treatments. Clinical trials currently recruiting participants include:
Community and Support
 Winnie
Winnie
"I was diagnosed officially with Junctional EB at 21, after the birth of my first son. It was actually a journey to get diagnosed because I was born and raised in the Philippines."
Winnie's Story Hodges
Hodges
"I want to encourage everyone one with EB to live life to the fullest - don't let it stop you from doing whatever you want to do."
Hodges' Story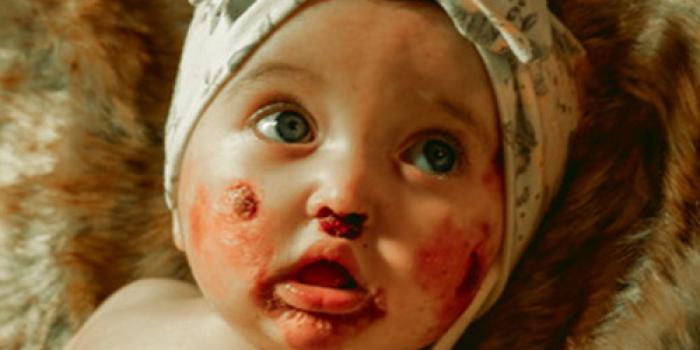 Caroline Georgia
Caroline Georgia
"We left the hospital within 48 hours after delivery with only her fingers being raw and a raspy faint cry. "
Caroline Georgia's StorySupport Resources
No one should face Dystrophic EB alone. debra of America offers free programs, personalized support, and trusted online resources to help individuals and families navigate life with EB.
Whether it’s finding answers to everyday challenges, accessing free wound care supplies, or connecting with others who understand—debra is here, every step of the way.
Visit our Get Help section to explore our free supportive programs, and check out our How To section for trusted, practical care guides.
Credit: C. Has et al, “Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility”, British Journal of Dermatology, October 2020