Dystrophic Epidermolysis Bullosa (DEB) is a subtype of Epidermolysis Bullosa characterized by extreme skin fragility affecting the deeper dermal layer beneath the epidermis.
Even minor friction or everyday activities can lead to painful blistering, chronic wounds, and significant scarring. Over time, individuals may experience joint contractures, loss of mobility, and reduced hand function. DEB typically presents at birth and may also affect mucous membranes and internal organs, which may result in serious medical complications and reduced life expectancy in the most severe forms.
While DEB is a lifelong condition, advances in clinical care and emerging treatments—including three FDA-approved therapies—are offering new hope for improving outcomes and quality of life. Navigate to a specific section by clicking the links below:
"My daughter, Addie, was born with skin missing on her feet and had to spend two weeks in the NICU undergoing tests because we didn’t know what was wrong. She was eventually diagnosed with Recessive Dystrophic Epidermolysis Bullosa (RDEB)."
Katelynn, Mom to Addie, Living with Recessive Dystrophic EB (RDEB)
Signs and Symptoms
Dystrophic Epidermolysis Bullosa (DEB) is characterized by extremely fragile skin that blisters from even minimal friction or everyday activities, affecting the deeper layers of the skin. DEB may be inherited as a dominant or recessive trait; generally, RDEB is more severe than dominant disease (DDEB); however, there is considerable phenotypic overlap between types. Hallmark signs and symptoms of DEB include:
- Chronic Blistering and Wound Formation: Blisters form in the deeper skin layers and mucous membranes, leading to non-healing wounds and increased risk of infection.
- Scarring and Mobility Issues: Scarring, joint contractures, and webbing can restrict movement and impair hand function.
- Mucosal Involvement: Blistering can affect the mouth, esophagus, and eyes, causing difficulties with swallowing, eating, and vision.
- Chronic Anemia and Other Complications: Ongoing blood loss from wounds can lead to anemia, further weakening the body and complicating recovery.
- Internal Organ Impact: In severe forms, internal organs like the gastrointestinal system and kidneys may be affected, contributing to further complications.
Subtypes of Dystrophic EB
Dystrophic Epidermolysis Bullosa is classified into several subtypes, each with its own pattern of blistering and severity. These subtypes are typically distinguished by the specific genetic mutations involved, the level of skin affected, and additional clinical features or symptoms. Below are the main subtypes of Dystrophic EB:
Recessive Dystrophic EB (RDEB)
RDEB is characterized by a wide spectrum of severity, from localized blistering to more widespread and debilitating skin involvement. It is typically divided into three subtypes based on the severity and extent of the condition: Localized, Intermediate, and Severe. RDEB is a lifelong condition, and the severity of the disease often dictates treatment and management strategies.
Localized RDEB
Specific Features
Restricted primarily to the hands, feet, or pretibial areas (shins). May present at birth or develop later.
Skin in affected areas is fragile, leading to recurrent blistering with minimal trauma.
Mild scarring and nail dystrophy (nail loss or deformities) often occur in the affected regions.
Rare or mild involvement of oral or esophageal areas.
Risk for squamous cell carcinoma (SCC) exists but is much lower compared to more severe forms, and tends to develop later in life.
Intermediate RDEB
(Previously known as RDEB generalized intermediate, non-Hallopeau-Siemens RDEB)
Specific Features
Widespread blistering, scarring and milia from birth or early childhood, with a predilection for bony prominences including the elbows, knees, ankles and dorsa of the hands and feet.
Mild flexion contractures (tightening of joints) and a striate-pattern of keratoderma (thickened skin) on the fingers may occur. Limited to moderate fusion of fingers or toes may develop.
The risk of squamous cell carcinoma (SCC) is increased in intermediate RDEB, but tending to occur later in adulthood.
Blistering, erosions, and scarring of the oral mucosa are possible. Progressive strictures can severely restrict nutritional intake.
Recurrent blistering and fissuring around the anal margin are frequent, contributing to pain and difficulty during defecation. Constipation is exacerbated by pain and reduced intake of fiber-rich foods.
The conjunctiva and cornea are often affected, leading to corneal erosions, scarring, pannus (clouding of the cornea), symblepharon (adhesion of the eyelid to the eye), and reduced visual acuity.
Nail dystrophy or loss due to trauma is common
Nutritional issues arise from reduced intake caused by factors such as microstomia (small mouth opening), dental caries, and esophageal strictures. The increased metabolic demands from chronic wounds, infection, and inflammation contribute to nutritional challenges.
Severe RDEB
(previously RDEB generalized severe, Hallopeau-Siemens RDEB)
Specific Features
Widespread and extensive blistering and skin fragility present from birth, often with areas of missing skin, especially on limbs (congenital skin ulceration).
Persistent, non-healing wounds at sites of repeated blistering. These contribute to chronic inflammation, infection, and tissue breakdown.
Extensive scarring leading to flexion contractures of large joints and pseudosyndactyly (digital fusion), resulting in classic mitten deformities of hands and feet.
Blistering and scarring may be more pronounced over pressure points like elbows, knees, ankles, and the dorsa of hands/feet.
Pigmented, wart-like skin lesions that may develop over time.
Scarring alopecia (hair loss) and scalp crusting are common and worsen with age.
Fragile nails prone to trauma, often lost progressively within the first few years of life.
Severe blistering, erosions, and scarring of the oral mucosa. May lead to microstomia (small mouth opening), ankyloglossia (tongue tethering), dental overcrowding and caries
Blistering and progressive scarring cause strictures that impair swallowing and nutrition, requiring regular surgical dilation.
Recurrent blistering around the anal margin leads to painful defecation.
May occur due to mucosal scarring.
Chronic constipation, possible dysmotility, and pain contribute to further nutritional challenges.
Blistering and inflammation of the conjunctiva and cornea, leading to corneal erosions, pannus formation, symblepharon (adhesions), scarring and visual impairment
Due to microstomia, dental issues, esophageal disease, chronic wounds, and high metabolic demand. This can lead to failure to thrive, weight loss, and growth delay.
A mixed type due to iron deficiency, chronic blood loss from wounds, and inflammation.
Poor bone density common, caused by reduced mobility, chronic inflammation, vitamin D and calcium deficiencies, pubertal delay.
Increased risk of fractures, including vertebral compression fractures.
Short stature and delayed puberty are frequently observed.
SCC is aggressive and the leading cause of death in severe RDEB, often develops during adolescence or early adulthood, with increasing incidence from teen years onward.
Can include post-streptococcal glomerulonephritis, renal amyloidosis, IgA nephropathy and progression to renal failure.
Rare but possible as part of long-term systemic disease.
Dominant Dystrophic EB (DDEB)
DDEB typically presents with a range of severity, from localized blistering to more widespread skin involvement. It follows an autosomal dominant inheritance pattern, meaning that an individual needs only one copy of the affected gene from either parent to inherit the condition. This type is generally less severe than Recessive Dystrophic EB (RDEB), but it can still cause significant complications and requires lifelong management. DDEB is categorized into two subtypes—Localized and Intermediate—based on the extent and severity of symptoms.
Localized DDEB
Specific Features
Often restricted to the hands, feet, or pretibial area (shins). May appear at birth or develop later in life, often after minor trauma.
Fragile skin in localized areas leads to frequent blistering and chronic wounds in affected regions.
Nail loss or deformities are common, especially with trauma or repetitive injury to the nails.
Mild scarring in localized areas, especially in places prone to friction or pressure, such as the hands and feet.
Squamous cell carcinoma (SCC) is possible but less, typically developing later in life.
Rare or mild. Mucosal blistering in the mouth and esophagus may occur.
Intermediate DDEB
Specific Features
Blistering tends to affect larger areas of the body, particularly over bony prominences such as elbows, knees, ankles, and the backs of hands and feet.
Scarring occurs in affected areas and is often accompanied by small white cysts (milia). The skin may become thicker and more fragile over time.
Mild contractures, keratoderma (thickened skin) in a striate pattern on the fingers, and limited webbing or fusion between digits may develop.
Blistering, erosions, and scarring may affect the mouth and esophagus, leading to nutritional challenges, strictures, or difficulty swallowing.
SCC can develop, tending to occur later in adulthood.
The conjunctiva and cornea can be affected, leading to erosions or scarring.
Nails may be damaged or lost, especially following trauma or blistering.
Recurrent fissures or blistering around the anal area can cause pain and constipation. GI involvement can also lead to nutritional challenges.
Other Dystrophic EB Variants
In addition to the more common forms of Dystrophic Epidermolysis Bullosa, there are several less common and distinct subtypes that present with unique challenges
Noted Clinical Symptoms
- Usually presents initially as localized or intermediate DDEB or RDEB in childhood and early adulthood.
- Characterized by intensely pruritic, excoriated violaceous papules or linear plaques and scars particularly on the lower legs, thighs and arms which can become more progressive from adolescence through adulthood.
- Nail dystrophy and milia are common.
- May co-exist with non-pruriginosa DEB within families.
Genetics
- Autosomal dominant or autosomal recessive inheritance.
- Monoallelic or biallelic mutations in COL7A1 similar to those identified in non-pruriginosa DDEB or RDEB without identification of a specific genotype-phenotype correlation for the pruriginosa phenotype.102–104
syn. bullous dermolysis of the newborn
Noted Clinical Symptoms
- Skin blistering presents at or shortly after birth, usually on the extremities.
- Aplasia cutis of the lower limbs may be present.
- Scarring and milia occur at sites of blistering.
- Skin fragility improves spontaneously and may resolve completely over the first year or two of life although nail dystrophy, particularly of the toenails, may persist throughout life.
Genetics
- Autosomal dominant or autosomal recessive.
- COL7A1 missense mutations, especially glycine substitutions, are most commonly associated but in-frame deletion, premature termination codon and alternative splicing mutations also described.
- Characteristic intra-epidermal retention of type VII collagen on immunohistochemistry and stellate bodies in dilated rough endoplasmic reticulum on transmission electron microscopy. These changes tend to improve in parallel with phenotypic improvement.
Noted Clinical Symptoms
- In the neonatal period and childhood skin blistering is usually generalized and of intermediate severity.
- From adolescence to early adulthood, a predilection for flexural sites develops, specifically in the axillae, groins, perianal area and natal cleft. In women, there may be marked vulvovaginal and inframammary skin blistering.
- Mucosal disease with blistering and scarring in the mouth and oesophagus is characteristic.
- Involvement of the external auditory canal may lead to narrowing or complete occlusion.
- Nail involvement is common.
Genetics
- Autosomal recessive inheritance.
- Usually results from compound heterozygosity for a loss-of-function COL7A1 mutation in combination with a missense mutation; specific glycine and arginine substitutions have been suggested as causative for this specific phenotype.
Noted Clinical Symptoms
- Skin fragility usually presents at or shortly after birth but may rarely be of late onset in adulthood.
- Blistering is limited in extent; it may affect predominantly the hands and feet, but in others may be restricted to the pretibial area.
- Nail dystrophy and loss are common.
- Oral and esophageal mucosal involvement are usually mild or absent.
Genetics
- Autosomal recessive inheritance.
- Compound heterozygosity for COL7A1 missense, splice site and frameshift mutations described.
Noted Clinical Symptoms
- Severe skin and mucosal fragility presenting from birth indistinguishable from severe RDEB.
- May have a family history of DDEB in family members.
Genetics
- Compound heterozygosity for a dominant COL7A1 glycine substitution mutation and a recessive mutation on the second allele.
Causes & Inheritance
Both RDEB (Recessive Dystrophic Epidermolysis Bullosa) and DDEB (Dominant Dystrophic Epidermolysis Bullosa) are genetic disorders caused by mutations in the COL7A1 gene, which provides instructions for producing type VII collagen, a crucial protein for skin integrity. These mutations result in defects in the skin’s ability to adhere to underlying tissues, leading to extremely fragile skin and blistering. However, the inheritance patterns and severity of symptoms differ between the two types.
The inheritance pattern of Recessive Dystrophic EB is autosomal recessive. This means an individual must inherit two mutated copies of the COL7A1 gene—one from each parent—to develop the condition. Both parents must be carriers of a mutated COL7A1 gene. In cases of RDEB, mutations can cause a complete or partial absence of type VII collagen, leading to widespread skin blistering and other severe complications.
The Inheritance Pattern of Dominant Dystrophic EB is autosomal dominant. In DDEB, only one mutated copy of the COL7A1 gene is necessary to develop the condition. This means that an individual with DDEB has a 50% chance of passing the condition on to each of their children. DDEB is usually inherited from an affected parent who carries one mutated copy of the gene. However, in some cases, the mutation can occur spontaneously as a new mutation in an individual with no prior family history of the condition.
Since one copy of the gene is typically functional in DDEB, it usually produces enough type VII collagen to maintain a certain level of skin adhesion. The mutation may result in a partial reduction in collagen production, but it doesn’t completely abolish it. In RDEB, the individual inherits two defective copies of the COL7A1 gene—one from each parent. This results in the complete or near-complete absence of type VII collagen.
Impact on Daily Life
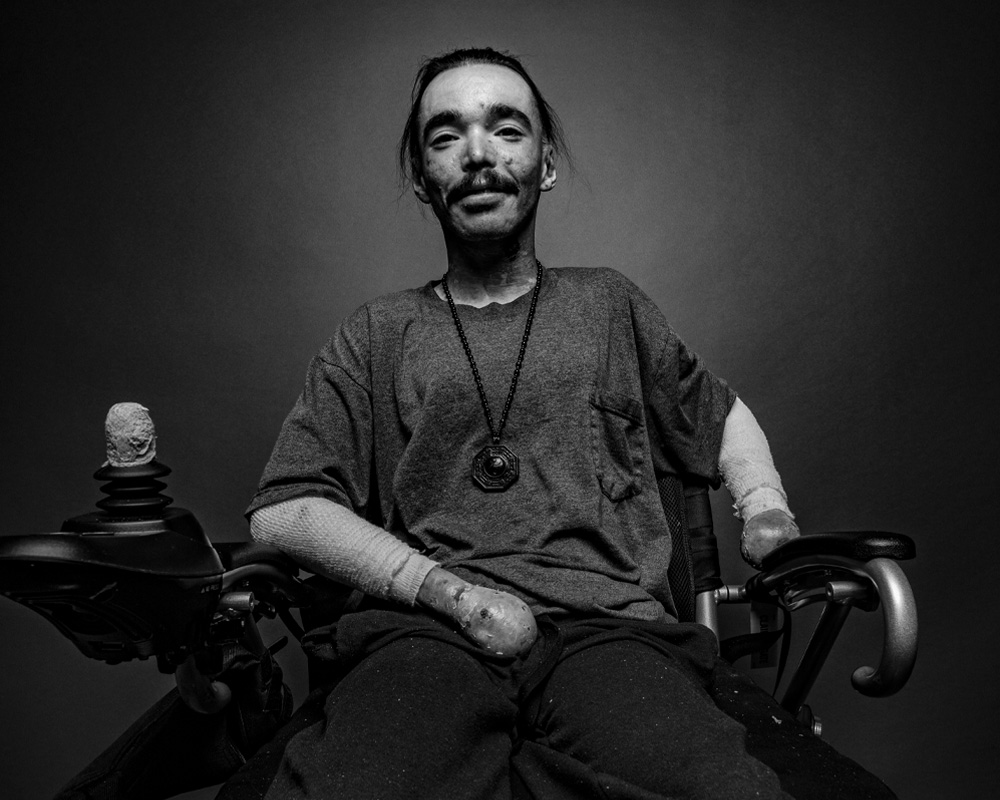
Living with DEB means navigating a condition that affects nearly every aspect of daily life. Frequent bandage changes, wound care, and constant vigilance are required to manage chronic skin fragility—blisters can form from minor friction or trauma, everyday tasks, or even spontaneously. Simple movements, like reaching or walking, can lead to blistering and skin tears, while painful joint contractures and scarring may limit mobility and require wheelchairs or other adaptive tools.
Everyday routines must be carefully adapted, and tasks others take for granted—like eating, dressing, or attending school—can take extra time, energy, and planning. Oral and esophageal involvement may make eating difficult or painful, sometimes requiring modified diets or feeding tubes to ensure proper nutrition.
Managing DEB often involves substantial adjustments at school, work, and home, as well as emotional resilience in the face of daily pain and limitations. Ongoing monitoring for complications—such as skin infections, internal organ issues, and aggressive squamous cell carcinoma (SCC)—is essential. Despite these challenges, many individuals find strength in community support, specialized care, and medical advancements that improve comfort and extend quality of life.
In the past three years, three FDA-approved treatments have been approved, most recently in April 2025, marking a historic turning point in DEB care and offering new hope for improved healing, reduced symptoms, and better long-term outcomes. The impact of DEB can vary widely depending on severity and presentation, with each person’s experience with EB being unique. Continue to explore our website for more detailed information on care and resources, and visit EBConnect.org, our online community platform, for valuable guidance and peer support.
Treatment and Care
Medical management of Dystrophic EB is largely supportive, focusing on wound care, pain management, and minimizing friction or trauma to the skin. Learn more about EB care strategies and practical information on our How To Care section.
Healthcare professionals can also refer to our Emergency Management page for clinical guidance in urgent situations.
Treatments
In an incredible achievement, there are currently three FDA approved for DEB.
-
FILSUVEZ®, developed by Chiesi Global Rare Diseases, is a prescription topical gel that supports wound healing in adults and children 6 months of age and older.
-
VYJUVEK™ (beremagene geperpavec-svdt) (VYEJOO-VEK), developed by Krystal Biotech, is the first FDA-approved treatment to address the genetic cause of Dystrophic Epidermolysis Bullosa (DEB). It is a topical gel that delivers new COL7A1 genes directly to DEB skin wounds to promote wound healing.
-
ZEVASKYN™ (prademagene zamikeracel), developed by Abeona Therapeutics, is the first and only autologous cell sheet-based gene therapy indicated for the treatment of wounds in adult and pediatric patients with Recessive Dystrophic Epidermolysis Bullosa (RDEB). Made from the patient’s own skin cells that have been gene-modified to express functional type VII collagen, ZEVASKYN sheets help address the genetic cause of RDEB in treated wounds.
Clinical Trials
Clinical trials play a critical role in advancing the understanding of Dystrophic EB and developing new treatments. Clinical trials currently recruiting participants include:
- Phase 3 Clinical Trial of D-Fi for DEB
- Natural History Study of DEB Eye Complications
- Phase 1/2A Clinical Trial of AGLE-102 for RDEB
- Phase 3 Clinical Trial of ABCB5+ Stem Cells for RDEB
- Phase 1 Clinical Trial of Dupilumab for All EB Types
- Research Study: Developing a Novel Artificial Intelligence Patient App to Recognize SCC in RDEB
- Phase 1/2 Clinical Trial of Rigosertib for RDEB with SCC
Community and Support
 Sarah
Sarah
"Living with EB comes with its unique challenges, but we have yet to face an obstacle we can’t overcome."
Sarah's Story Nicole & Hayden
Nicole & Hayden
"Learning about our son’s diagnosis was very challenging to say the least. We had a lot of trial & error to make Hayden as comfortable as possible. "
Nicole and Hayden's Story"I was diagnosed with Dominant Dystrophic EB (DDEB) at 18 months old. Growing up with EB came with many hardships "
Kaylyn's StorySupport Resources
No one should face Dystrophic EB alone. debra of America offers free programs, personalized support, and trusted online resources to help individuals and families navigate life with EB.
Whether it’s finding answers to everyday challenges, accessing free wound care supplies, or connecting with others who understand—debra is here, every step of the way.
Visit our Get Help section to explore our free supportive programs, and check out our How To section for trusted, practical care guides.
Credit: C. Has et al, “Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility”, British Journal of Dermatology, October 2020